原创 朱加伟、宫蕾 研之成理 2023-03-20 10:24 发表于浙江
▲第一作者:宫蕾;朱加伟
通讯作者:木士春 通讯单位:武汉理工大学 论文DOI:10.1021/acscatal.2c06340
01 全文速览
本工作借助Mn单原子位点与Pt活性物质间的强相互作用,有效调制了Pt物种的粒径大小,提升活性原子利用率。此外,电子由单原子Mn位点转移至邻接的Pt位点上,使得富电子Pt位点的d带中心负移,削弱了对关键中间体的表面吸附,从而提升了催化剂的本征氧还原(ORR)及析氢(HER)活性。
02 背景介绍
A.氢燃料电池涉及的催化问题氢能是实现“双碳”目标的重要能源载体。氢燃料电池可直接将氢能转化成化学能,具有高的能量转换效率和清洁性优势,是未来重要的能源转换系统。但氢燃料电池受其缓慢的阴极氧还原反应(ORR)动力学限制,且其阳极需要可持续的绿氢燃料输入。因而,设计制备合理的催化剂加速ORR和析氢反应(HER)催化是该领域的研究重点。对这两个催化反应而言,碳载铂(Pt/C)催化剂是目前最高效的商业催化剂。从以往的相关研究来看,Pt基催化剂展示出更低的过电位、快速的反应动力学,在ORR/HER催化反应中具有一定性能优势。但Pt金属储量低、价格高、稳定性较差,难以在能源装置中大规模应用。因此,进一步增强Pt基催化剂的本征活性、提高Pt金属的利用率、降低用量,并提升其稳定性,是解决该问题的关键。
B.单原子催化体系由于发达的孔隙结构和可调制的金属种类、配位结构,金属-有机框架(MOFs)材料及其衍生碳基纳米材料可为电催化提供不同的活性中心,已被广泛开发应用于各类电催化反应中。在MOFs框架基础上,可经由不同构筑策略获得单原子催化剂。其中,最普适的策略是借助MOFs孔隙结构的空间限制效应有效隔离金属位点,在空间上实现单原子的高度分散。对不同的金属中心,相较普遍研究的Fe/Co单原子,Mn单原子对ORR反应尽管具有较低的催化活性,但Fenton副反应和两电子选择性被显著抑制。因而,向单原子Mn-N-C体系中引入低载量的贵金属活性物种,有望提高整体催化剂活性的同时保持高的稳定性。
03 研究出发点
首先,为了解决单原子Mn-N-C催化剂的活性问题,拟将高本征活性的Pt活性物种引入到体系中。为了提高催化剂的整体效益,可调节前驱体用量实现超低的Pt载量。此外,由于活性金属物质与M-N-C骨架间存在强金属-载体效应(SMSI),Pt物种与单原子Mn-N-C位点间会存在强相互作用,可有效抑制表面Pt物种的团聚,有望获得最佳颗粒粒径分布(Nano Energy 2021, 88, 106221;J. Energy Chem. 2022, 65, 48-54)。最后,Pt与单原子Mn位点间的电荷密度重排则可进一步优化活性位的电子结构,使其对ORR/HER反应关键中间体的吸附得到优化,获得更高的本征催化活性。
04 图文解析
A.材料合成
通过逐级构筑策略制备了具有多活性中心的Pt@Mn-SAs/N-C催化剂。首先是制备Zn/Mn-ZIF前驱体。在热解过程中,借助ZIF的孔隙结构限制Mn原子的迁移与团聚,获得了单原子Mn-SAs/NC材料。接着,将其与PtCl62-溶液进行反应。带正电的Mn位点与带负电的PtCl62-发生静电吸引,并利用孔隙结构直接捕获住PtCl62-,获得PtCl62-@Mn-SAs/N-C。在还原性气氛下进行热还原,获得最终产物Pt@Mn-SAs/N-C。
从电镜结果可以看出,前驱体与热解产物均呈现菱形十二面体形貌。N-C仅表现出无定形乱层碳特征,Mn-SAs/N-C中可观测到明确的单原子Mn位点,而在Pt@Mn-SAs/N-C中则可观察到Mn单原子位点与尺寸在2.43 nm左右的Pt纳米颗粒的共存。这表明,由于单原子Mn位点对Pt前驱体的强锚定作用,以及孔隙结构的限制效应,Pt物种的尺寸获得明显约束。ICP-OES结果则表明,Pt物种的整体引入量仅为1.98 wt%。综上所述,在超低Pt负载量的前提下,利用强相互作用获得了较低的颗粒尺寸与高的活性原子利用率。

▲Figure 1. (a) Schematic of the preparation process for Pt@Mn-SAs/N-C. (b) Scanning electron microscopy (SEM) image and (c) transmission electron microscopy (TEM) image of Mn-SAs/N-C. (d) SEM image, (e) TEM image, (f) high-resolution transmission electron microscopy (HRTEM) image, and (g) aberration-corrected scanning transmission electron microscopy (ac-STEM) image of Pt@Mn-SAs/N-C. (h-l) high-angle annular dark field (HAADF) image and relevant elemental mapping of Pt@Mn-SAs/N-C.
B.物性表征
XRD结果进一步确认Pt@Mn-SAs/N-C中Pt物相的存在。从XPS精细谱分析结果来看,引入Pt物种后,Pt@Mn-SAs/N-C的Mn 2p谱图相较Mn-SAs/N-C出现明显的正移趋势。这表明单原子Mn位点上存在电子损失。同样地,Pt@Mn-SAs/N-C的Pt 4f谱图相较Pt@N-C表现出负的峰位移,表明Pt位点上的得电子特征。上述结果证明,单原子Mn位点与引入的Pt物种间存在强耦合作用,电子由Mn位点向邻接的Pt位点转移。
从更精确的XAS分析结果可进一步确认Pt@Mn-SAs/N-C中占主导地位的Mn-N配位构型(1.62 ?)。其平均配位数约为4,为常规的Mn-N4结构。而在2.36 ?处的小峰归属于Mn-Pt配位,表明单原子Mn-N4位点与所锚定的Pt物种的强耦合成键作用。而Pt@Mn-SAs/N-C中的Pt主要以Pt-Pt配位存在,配位数为4.5,是典型的Pt纳米颗粒特征。上述光谱分析揭示了Pt@Mn-SAs/N-C中的价键结构特征。

▲Figure 2. (a) XRD patterns of Pt@Mn-SAs/N-C and Mn-SAs/N-C. (b) Mn 2p spectra of Pt@Mn-SAs/N-C and Mn-SAs/N-C. (c) Pt 4f spectra of Pt@Mn-SAs/N-C. (d) X-ray absorption near-edge structure (XANES) and (f) extended X-ray adsorption fine structure (EXAFS) spectra of Mn K-edge for Pt@Mn-SAs/N-C, Mn foil, and Mn2O3. (e) XANES and (g) EXAFS spectra of Pt L3-edge for Pt@Mn-SAs/N-C, Pt foil, and PtO2. WT-EXAFS of Mn K-edge for (h) Pt@Mn-SAs/N-C, (i) Mn foil, and (j) Mn2O3.
C.电催化性能
对所制备的催化剂在酸、碱性介质中的ORR及HER性能进行分析。其中,在酸性ORR测试中,Pt@Mn-SAs/N-C具有极佳的ORR催化活性,其半波电位约为0.896 V,远高于对比样和商业Pt/C催化剂(0.85 V)。低载量Pt活性物种与单原子Mn位点的协同电子相互作用显著增强了催化剂的整体ORR活性。由于低的Pt载量,Pt@Mn-SAs/N-C催化剂在0.85 V和0.9 V下的质量活性分别为0.62 and 0.39 A mgPt-1,是Pt/C催化剂的9.6和11.1倍,展示了很好的经济效益。而稳定性测试则表明,Pt@Mn-SAs/N-C催化剂在5000圈CV循环前后半波电位仅衰减了1 mV,并且在i-t测试中也具有很小的电流密度衰减。此外,Pt@Mn-SAs/N-C催化剂还具有很好的抗甲醇性能。上述结果表明,对于酸性ORR反应,Pt@Mn-SAs/N-C催化剂是商业Pt/C催化剂理想的替代催化剂。
此外,Pt@Mn-SAs/N-C催化剂在碱性ORR、酸性HER、碱性HER反应中均表现出很好的催化活性、高的质量活性及稳定性。
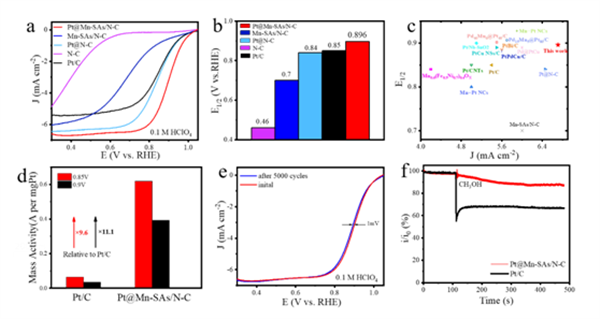
▲Figure 3. (a) ORR performance in acidic media. (a) LSV curves and (b) half-wave potentials of Pt@Mn-SAs/N-C, Mn-SAs/N-C, Pt@N-C, N-C, and commercial Pt/C catalysts in 0.1 M HClO4 solution. (c) Acidic ORR performance comparison of Pt@Mn-SAs/N-C with other reported catalysts, (d) Mass activities of Pt@Mn-SAs/N-C and Pt/C catalysts at 0.85 V and 0.9 V. (e) Acidic LSV curves of Pt@Mn-SAs/N-C before and after 5000 cycles and inserted chronoamperometry test of Pt@Mn-SAs/N-C and Pt/C catalysts. (f) Methanol tolerance tests of Pt@Mn-SAs/N-C and Pt/C catalysts in acidic media.
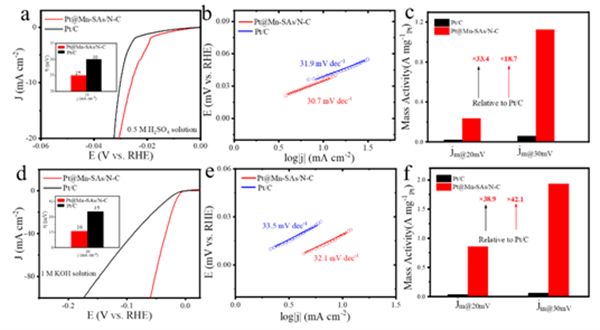
▲Figure 4. (a) LSV curves and (b) corresponding Tafel slopes of Pt@Mn-SAs/N-C and Pt/C catalysts in acidic media. (c) Mass activity of Pt@Mn-SAs/N-C and Pt/C at 20 and 30 mV in 0.5 M H2SO4 solution. (d) LSV curves and (e) corresponding Tafel slopes of Pt@Mn-SAs/N-C and Pt/C catalysts in alkaline media. (f) Mass activity of Pt@Mn-SAs/N-C and Pt/C at 20 and 30 mV in 1 M KOH solution.
D.理论计算
为了进一步研究Pt物种与单原子Mn位点间的相互作用,进行了密度泛函理论(DFT)计算。在ORR及HER吉布斯自由能谱图中,Pt@Mn-N4模型表现出最低的反应能垒。这表明,Pt与单原子Mn的耦合可有效降低活性金属原子位点上的理论ORR/HER催化活性。
理论催化活性的提升主要是由于复合模型在费米能级上的电子态的提升。这有利于提高整体催化剂的本征电子传导作用。差分电荷密度分析则进一步揭示了耦合界面上由单原子Mn-N4位点向相邻的Pt位点的电子转移,这与XPS分析结果一致。所导致的富电子Pt位点展现出了负移的d带中心,削弱了Pt位点与关键OH*和H*中间体的吸附作用,从而降低了ORR/HER的反应能垒。

▲Figure 5. (a) ORR free energy diagram of Mn-SAs/N-C, Pt@Mn-SAs/N-C, and Pt models. (b) HER Gibbs free energy diagrams of these theoretical models. (c) As-constructed model and differential charge density of Pt@Mn-SAs/N-C. (d) Partial density of states (PDOS) of the d orbital of Pt atoms in Pt@Mn-SAs/N-C and Pt models.
05 总结与展望
作者通过向Mn-N-C单原子体系中引入额外的活性Pt物种。在保留反应选择性和稳定性优势的同时,进一步提升了催化性能。由于Pt与Mn单原子间的强耦合作用,有效限制了Pt的过度生长,在低载量的前提下提升了活性原子的利用率。所制备的Pt@Mn-SAs/N-C催化剂对酸性及碱性介质下的ORR和HER反应均表现出优异的催化活性及稳定性。本征活性的提升主要源自于Mn-N4位点与Pt物种间的电荷密度重排,使得富电子Pt位点的d带中心负移,削弱了关键OH*和H*中间体的吸附,导致反应能垒降低。该工作为ORR和HER催化提供了一种高效稳定的催化剂候选,为研究多活性组分的催化体系提供了研究范式。
06 作者介绍
木士春教授,武汉理工大学首席教授,博士生导师,国家级高层次人才。长期致力于电解水制氢和质子交换膜燃料电池催化剂研究。以第一作者或通讯作者在 Nat. Commun.、Adv. Mater.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Energy Environ. Sci.等国内外期刊上发表300余篇高质量学术论文。课题组网站:http://www.polymer.cn/ss/shichunmu/index.html原文链接:https://pubs.acs.org/doi/10.1021/acscatal.2c06340