
研究背景
氧还原反应(ORR)作为质子交换膜燃料电池(PEMFC)和金属-空气电池等清洁能源转换装置的核心反应,其反应速率决定了装置的工作效率。迄今为止,Pt基催化剂在酸性和碱性介质中具有最高的催化ORR动力学活性,但由于Pt的价格昂贵以及抗甲醇毒化性较差,极大地限制了Pt基催化剂在这类装置中的广泛应用。因此,选用廉价、活性高、耐久性优异的电催化剂来替代Pt基催化剂一直以来都是热门话题之一。
成果介绍武汉理工大学的木士春教授、郑州大学的张佳楠教授等人合理设计了双金属原子分散的催化剂(Fe,Mn/N-C),相邻原子分散的Mn-N能有效激活FeIII位点,使FeIII在FeN4位点中实现单个eg电子填充(t2g4eg1),从而更加有利于电子进入O的反键轨道上,实现O2的活化。磁化率测试与理论计算共同表明,相邻原子分散的Mn-N可以通过自旋态转变、电子调制激活FeIII位点,使得Fe,Mn/N-C在碱性和酸性介质中均具有优异的ORR活性与稳定性,在0.1 M KOH下半波电位为0.928 V、0.1 M HClO4下半波电位为0.804 V。将其作为阴极催化剂,组装锌空气电池时,峰值功率密度达160.8 mW cm-2,并能够稳定进行充放电循环。相关工作以“Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity”为题在Nature Communications上发表。
图文介绍
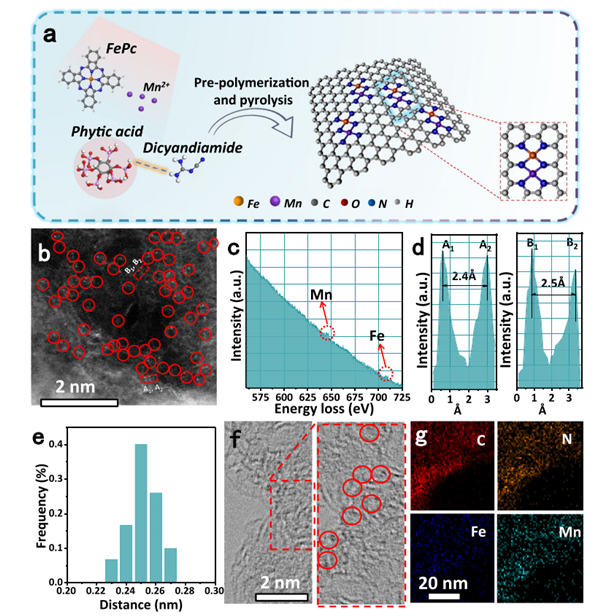
图1 Fe,Mn/N-C催化剂的合成示意图以及TEM表征
图1a为Fe,Mn/N-C催化剂的合成示意图,双氰胺作为C、N源,酞菁铁(FePc)与Mn(NO3)2分别作为金属前驱体,通过前聚合、热解过程,得到Fe,Mn/N-C催化剂。采用相同的方法合成了单金属原子分散Fe/N-C和Mn/N-C催化剂,作为对照组。从图1b的HAADF-STEM图像中,可以清晰地观察到一些亮点,表明Fe/Mn原子对随机分布在N掺杂碳的表面。通过EELS谱图(图1c)表明了Fe和Mn元素共存。此外,作者还对30多个原子对的间距进行了统计分析,图1d、e所示,发现双原子对的间距为0.25±0.02 nm。从HRTEM图像(图1f)可观察到明显的晶格畸变,这可能是由于Fe/Mn双原子与N的配位造成的。EDS元素映射分析进一步表明,C、N、Fe和Mn在Fe,Mn/N-C上均匀分布。

图2 XAS、57Fe M?ssbauer光谱以及催化剂的磁化率
通过XAS对Fe、Mn的局部配位结构进行分析,如图2a的Fe的K边XANES谱图所示,Fe,Mn/N-C、Fe/N-C的吸收近边处于FeO与Fe2O3之间,表明Fe的价态处于+2~+3之间。
FT-EXAFS谱图(图2b)表明Fe,Mn/N-C与Fe/N-C均在1.4 ?处出现相同的峰,对应Fe-N配位键。
此外,与Fe/N-C相比,Fe,Mn/N-C在2.16 ?处还有额外的峰,代表Fe-Metal配位键。这一结果与Mn的XAS测试结果相类似,如图2c、d所示,与Mn/N-C相比,Fe,Mn/N-C在2.34 ?处还有额外的峰,代表Mn-Metal配位键。通过结合理论计算,得出了这种新型的Fe,Mn-N6构型。
采用57Fe M?ssbauer分析对不同的Fe物种进行了鉴别。如图2f所示,对于Fe,Mn/N-C,D1峰具有较大的同质异能位移值与四极分裂值,可归属于类似FePc的FeII-N4物种。D3峰对应高自旋态N-(FeIII-N4)-N物种,由于构型稳定,不能作为催化活性位点。D4峰代表N-(FeIII-N4)物种的中间自旋态,不饱和的配位结构使得该物种能作为高活性的催化活性位点。定量分析表明,D1、D3、D4的含量分别为13.5%、27.1%、59.4%,证实了FeIII在Fe,Mn/ N-C中具有中间自旋态结构。通过零场冷却磁化强度测试,如图2g、h所示,计算了Fe、Mn/N-C和Fe/N-C的有效磁矩分别为3.75μeff、2.16 μeff,并由此推算未配对d电子数(n):Fe/N–C中n为1.3,表明FeIII离子在没有eg填充的情况下处于低自旋状态,没有电子占据σ*反键轨道,导致FeIII与O2的相互作用非常强,Fe4+-O22-键相当稳定。而在Fe,Mn/N-C中,n为3,受相邻低自旋MnIII的影响,Fe的eg上有单电子填充,从而赋予了催化剂较高的催化活性。
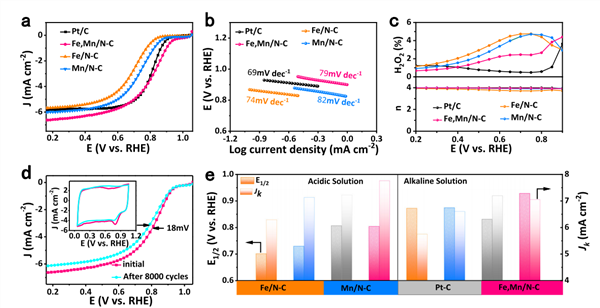
图3 ORR性能
在0.1 M HClO4下测试具有独特自旋结构的FeIII的ORR活性。如图3a的LSV曲线所示,Fe,Mn/N-C的半波电位高达0.804 V,与商业Pt/C催化剂接近,并远优于Fe/N-C、Mn/N-C以及大多数文献报道的催化剂的半波电位。
根据Tafel曲线,如图3b所示,Fe,Mn/N-C的Tafel斜率低至79 mV dec-1。
通过RRDE进一步考察转移电子数和H2O2产率,如图3c所示,n接近4,H2O2产率低于4%。
此外,Fe,Mn/N-C也表现出良好的稳定性,如图3d所示,经过8000次电位循环后,半波电位仅减小18 mV。
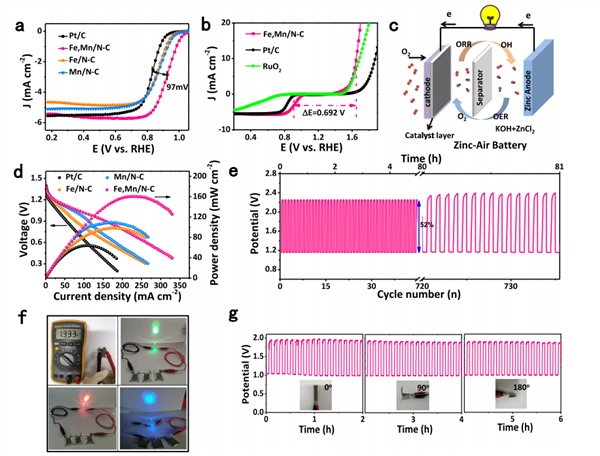
图4 锌空气电池性能
进一步在0.1 M KOH下测试Fe,Mn/N-C的ORR活性,如图4a所示,其半波电位高达0.928 V,高于商业Pt/C催化剂。通过ΔE进行评估氧电催化剂的双功能(ORR/OER)催化活性,如图4b所示,Fe,Mn/N–C具有最低的ΔE,为0.692V。
因此,作者将Fe,Mn/N–C催化剂组装到液态、柔性全固态可充电锌空气电池上。如图4c所示为液态可充电锌空气电池的装置示意图。图4d的极化曲线表明,锌空气电池的开路电压达1.4 V,峰值功率密度为160.8 mW cm-2。同时,在恒电流5.0 mA cm-2下进行循环充放电,如图4e所示,经过81h后充放电电压未明显变化,进一步证明了Fe,Mn/N–C催化剂的优异稳定性。在柔性全固态可充电锌空气电池上,其开路电压达1.33 V,三个电池串联时可稳定点亮不同颜色的LED灯泡,如图4f所示。同时,在恒电流1.0 mA cm-2下、不同弯曲角度下进行循环充放电,如图4g所示,电池充电电压、放电电压稳定保持在1.94 V、1.04 V。
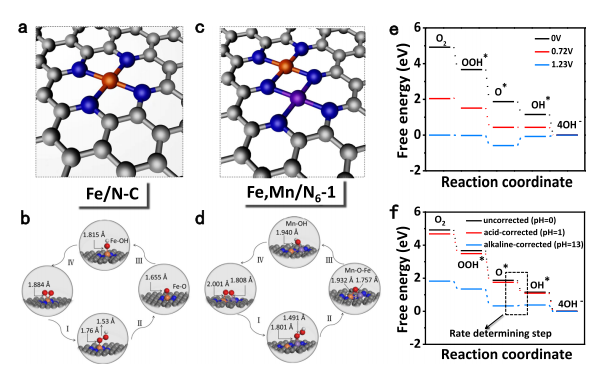
图5 DFT计算
采用DFT计算来阐明Fe,Mn/N-C的ORR活性来源。Fe/N–C、Fe,Mn/N–C的优化结构模型与反应模型如图5a-d所示,在Fe/N–C上,Fe–O2 键长为1.884 ?,与O2的过度结合导致后续的质子-电子转移步骤受限,从而易形成更多过氧化物中间体,降低了ORR活性和稳定性。而在Fe,Mn/N-C上,O2在Fe/Mn原子对的吸附结合得到优化,从而降低了解离能垒。如不同电位下的ORR自由能图所示,在U=0 V下,ORR的每个基元反应自由能均小于0。在U=0.72 V下,O*生成OH*步骤的反应自由能为0,而其他步骤的自由能小于0,此时将这个电位称为热力学极限电位。相比之下,Mn/N-C与Fe/N-C的热力学极限电位分别为0.42V、0.36 V。因此,在此条件下,Fe,Mn/N-C具有较高的ORR反应性,与实验结果一致。
此外,如图5e, f所示,在pH值为0时,酸校正后的自由能图没有明显变化,而碱校正后的自由能图有明显变化。然而,在相同的计算模型下,pH校正操作不会改变ORR的速率决定步骤。因此,虽然过电位的数值会发生变化,但由于pH校正值相同,不同模型的理论ORR趋势不会发生变化。也就是说,电解质的影响不会改变目前的结论:在所有模型中,Fe,Mn/N6模型的理论ORR活性最好,过电位最小。总结与展望本文合成了双金属原子分散的Fe,Mn/N-C催化剂,并以Fe-N/C、Mn-N/C催化剂为对照组,揭示了过渡金属离子3d轨道的自旋态结构对ORR活性的重要影响。实验结果与理论计算共同表明,相邻原子分散的Mn-N能有效激活FeIII位点,使FeIII在FeN4位点中实现单个eg电子填充(t2g4eg1),从而更加有利于电子进入O的反键轨道上,实现O2的活化。因此,Fe,Mn/N-C催化剂在酸性介质、碱性介质均表现出优异的ORR性能,在0.1 M KOH下半波电位为0.928 V、0.1 M HClO4下半波电位为0.804 V。
DFT理论计算进一步证实了电子结构调整后的Fe,Mn/N-C和含氧中间体具有适当的键长和键能,从而改善了ORR动力学。最后,将Fe,Mn/N-C应用于实际的液态、柔性全固态可充电锌空气电池中,同样表现优异的活性与稳定性。本工作为优化非贵金属催化剂,并实现催化剂在燃料电池、金属-空气电池和其他可再生能源系统的应用提供了新的见解。
文献信息
题目:Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. (Nat. Commun., 2021, DOI:10.1038/s41467-021-21919-5)
链接:https://www.nature.com/articles/s41467-021-21919-5