赵宏宇 研之成理 2025年05月13日 10:09 浙江
全文速览
本研究基于密度泛函理论(DFT)计算预测,通过采用温度调控的熔盐辅助合成策略,成功构建了由正交相和立方相RuTe2(m-RuTe2)组成的异相同质结催化剂。理论研究表明,该异相同质结构通过电荷平衡效应、优化的Ru-Te轨道杂化程度及氢吸附能。在碱性条件下,m-RuTe2在10 mA cm-2电流密度下仅需35 mV的析氢反应过电位,这一性能不仅显著优于单相RuTe2催化剂,更超越了商业Pt催化剂的活性。此外,得益于Ru-Te结构单元之间强烈的电子耦合作用,该催化剂有效抑制了Ru活性中心的溶解,表现出优异的长期稳定性。
背景介绍
氢气因其高能量密度和绿色环保特性,被视为极具发展潜力的可持续能源。在催化领域,与传统异质结构催化剂相比,异相同质结构催化剂展现出显著优势:其单一的化学组成确保了活性中心的均匀分布,从而显著提升了催化活性、稳定性和选择性。更重要的是,异相同质结构中两相间的功函数(WF)差异可诱导形成内置电场,这一特性不仅促进了电子转移和电荷再分布过程,还能优化活性中间体的吸附行为,最终实现催化性能的显著提升。
本文亮点
1、RuTe2异相同质结两相之间的功函数差诱导了内建电场的形成。
2、内置电场驱动的电荷转移和重排优化了RuTe2在反应中间体上的吸附。
3、采用温度调节熔盐辅助策略制备了异相同结结构RuTe2催化剂。
4、RuTe2异相同质结在碱性介质中表现出优异的析氢反应活性和长期稳定性。
图文解析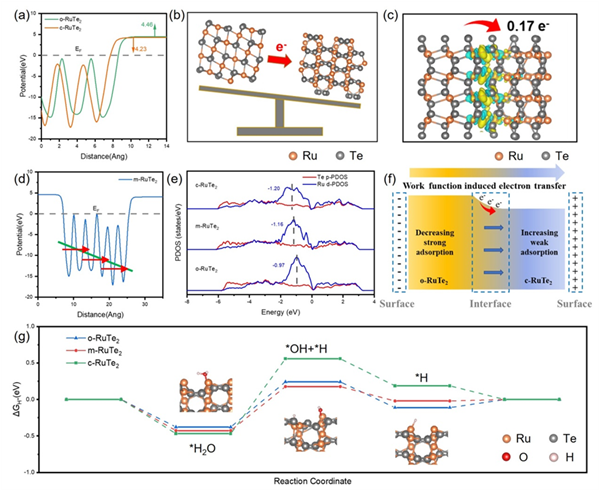
图1. (a)c- RuTe2和o- RuTe2的功函数。(b)功函数诱导的电子转移示意图。(c)m- RuTe2的差分电荷密度分布和Bader电荷分析。(d)m- RuTe2的功函数。(e)RuTe2中Ru、Te的分波态密度(PDOS)。(f)m- RuTe2中的电荷转移过程。(g)碱性HER的吸附自由能图
DFT理论计算结果揭示,通过构建正交相(o-RuTe2)与立方相(c-RuTe2)的异相同质结模型(m-RuTe2),可显著提升析氢反应(HER)活性。两相间显著的功函数差异诱导产生强的内置电场,该电场不仅驱动界面电子的定向转移和重新分布,还优化了反应位点的电子结构,从而促进了氢中间体的吸附-解离平衡。相较于单一晶相,m-RuTe2的异质界面在降低水解能垒的同时还优化了m-RuTe2表面的氢吸附自由能ΔGH*。这表明催化剂电子结构的优化可显著加速析氢反应动力学(图1)。

图2.(a)所有合成材料的XRD图谱。(b)所有合成材料的Ru 3p的XPS光谱。(c)所有合成材料的Te 3d的XPS光谱。分别从(d)o-RuTe2、(e)m-RuTe2和(f)c-RuTe2观察价电子的电子局域函数(ELF)的2D切片,数值是Ru和Te的计算Bader电荷值。
通过温度调控的熔盐辅助法,成功实现了不同RuTe2晶相的可控合成。XRD分析表明,500 ℃条件下合成的样品呈现正交相结构(o-RuTe2),700 ℃的产物为立方相(c-RuTe2);而在600 ℃这个中间温度下,样品显示出o-RuTe2和c-RuTe2两相共存的特征(m-RuTe2)。值得注意的是,XPS分析显示,随着合成温度从500 ℃升高至700 ℃,Ru与Te之间的电荷转移程度逐渐增强,同时Ru的氧化态也随之升高。进一步研究发现,与单一晶相的o-RuTe2或c-RuTe2相比,两相共存的m-RuTe2展现出独特的电子结构特征。这主要源于两相间功函数差异诱导产生的内建电场促进了界面电荷的转移与重排,从而形成了适中的Ru-Te轨道杂化程度。这种优化的电子结构为催化反应提供了有利条件。电子局域函数(ELF)分析进一步证实,从o-RuTe2到c-RuTe2存在逐步增强的电荷转移趋势,与实验结果一致。(图2)

图3. (a)o-RuTe2、(b)c-RuTe2和(c)m-RuTe2的TEM图像。(d-e)m-RuTe2的HRTEM图像和相应区域的FFT图案:橙色:o-RuTe2,红色:c-RuTe2。(f)m-RuTe2的EDS作图。(g)异相同质结结构形成示意图。
TEM图像分析表明,三种RuTe2样品呈现出显著不同的结构特征:o-RuTe2以球形纳米颗粒为主,c-RuTe2表现为典型的棒状纳米结构,而m-RuTe2则展现出独特的"球-棒"复合结构,即大量细小的棒状纳米颗粒生长于球形纳米颗粒表面。HRTEM图像进一步揭示了m-RuTe2中两相共存的结构特征,并观察到了从c-RuTe2向o-RuTe2的相变过渡区域。这一结果直接证实了RuTe2异相同质结构的成功构建。(图3)
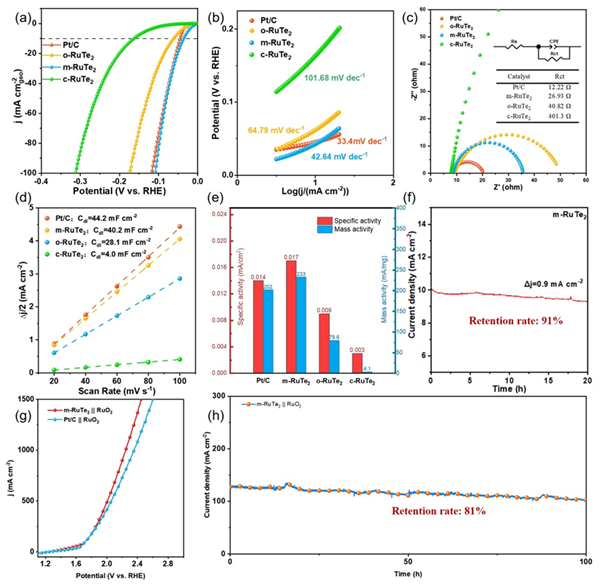
图4.(a)所有不同RuTe2样品的LSV曲线(扫描速率:10 mV s-1)。Pt/C用作参比。(b)每个样品的Tafel斜率和(c)奈奎斯特图。(d)各种催化剂的Cdl。(e)各种催化剂在50 mV过电位下的质量活性和比活性。(f)m-RuTe2的恒定电压稳定性测试。(g)m-RuTe2//RuO2和Pt/C//RuO2朝向整体水分解。(h)m-RuTe2//RuO2的恒定电压稳定性测试。
采用标准三电极体系在1.0 M KOH电解液中评估了不同RuTe2样品的析氢反应性能。其中,单斜相m-RuTe2表现出最优异的催化活性,仅需35.3 mV的过电位即可达到10 mA cm-2的电流密度,显著优于正交相o-RuTe2(62.3 mV)和立方相c-RuTe2(164.3 mV)。此外,m-RuTe2的反应动力学和电化学活性表面积(ECSA)均优于o-RuTe2和c-RuTe2,并展现出比商业Pt/C更优的质量活性和比活性。m-RuTe2在20小时的恒电流电解过程中,电流密度保持率达91%;经过2000次循环伏安(CV)加速老化测试后,其过电位仅增加2 mV,展现出出色的长期稳定性。在m-RuTe2||RuO2双电极体系中,在2.28 V的电压下即可驱动1000 mA cm-2的电流密度,优于商业Pt/C||RuO2体系性能(2.36 V)。同时,该体系还还表现出优良的长期稳定性。(图4)
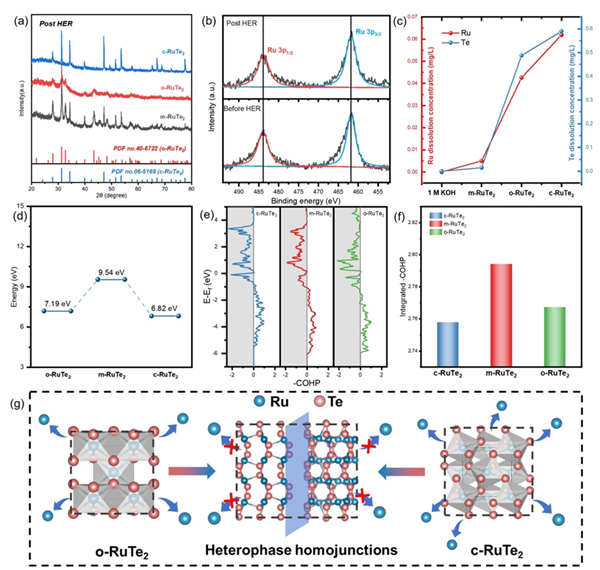
图5.(a)HER测试后所有合成材料的XRD图谱。(b)测试前后m-RuTe2的Ru 3p的XPS光谱。(c)不同RuTe2催化剂连续催化反应后溶解在电解质中的Ru和Te的浓度。(d)不同RuTe2催化剂中Ru的脱金属能量。(e)不同RuTe2催化剂中Ru-Te能带的-COHP(f)不同RuTe2催化剂中Ru-Te能带的积分-COHP至费米能级比较。(g)形成抑制反应性金属溶解的异相同质结结构的两相的示意图。
通过对比HER测试前后的XRD图谱,发现m-RuTe2的晶体结构保持完好,特征衍射峰无明显变化;XPS分析进一步证实其表面化学态保持稳定。ICP-OES定量分析显示,在20小时恒电位电解后,m-RuTe2中Ru和Te的溶解浓度低于o-RuTe2和c-RuTe2。这些测试结果表明,m-RuTe2异相同质结构具有增强的结构稳定性。DFT计算进一步揭示了该稳定性增强的机理:首先,m-RuTe2中Ru的溶解能垒高达9.54 eV,高于o-RuTe2和c-RuTe2;其次,晶体轨道哈密顿布居(COHP)分析表明,m-RuTe2具有最强的Ru-Te共价相互作用(-ICOHP = 2.79 eV)。这种增强的稳定性源于异相界面处内置电场驱动的电荷再分配与强化的Ru-Te共价耦合的协同效应:内置电场促进界面电子离域,而增强的共价作用则提供了结构骨架支撑,二者共同抑制了金属溶解,有效缓解了长时间HER过程中的结构退化问题(图5)。
总结与展望
本研究成功设计并构建了由立方相和正交相组成的异相同质结(m-RuTe2)催化剂。两相共存诱导的内置电场效应显著促进了电荷转移与重排过程,进而优化了活性中间体在催化剂表面的吸附行为,从而加速了催化反应的动力学进程。此外,Ru-Te之间的强电子耦合作用显著抑制了析氢反应过程中Ru活性位点的失活和溶解问题。实验结果显示,m-RuTe2异相同质结催化剂具有远优于单相RuTe2和商业Pt催化剂的催化活性和长期稳定性。这一重要发现不仅证实了异相同质结结构设计在提升传统单相催化剂性能方面的独特优势,还极大地拓展了催化剂的研究维度,为其他催化体系的材料设计提供了新的研究思路。