1. 嵌段共聚物非平衡态自组装行为及其机理
嵌段共聚物体系在纳米尺度上形成的有序结构是获取新型高分子材料所需要的聚集态结构的重要来源之一。但是,由于线形AB两嵌段共聚物体系能够形成的稳定纳米有序结构数目十分有限,而采用多组分嵌段共聚物体系和更为复杂的高分子链拓扑结构来获取所需要的稳定纳米有序结构又需要十分复杂的化学合成过程,不利于成本的降低,因此,如何调控简单嵌段共聚物体系的非平衡态结构演化动力学过程来获取所需要的纳米有序结构(非平衡态过程控制自组装)就变得十分具有吸引力。其中的关键在于如何构建简单嵌段共聚物体系中的不稳定状态,从而使由构建的不稳定状态出发的非平衡态结构演化动力学过程最终到达人们所需要的纳米有序结构。尽管由非平衡态结构演化动力学过程俘获的纳米有序结构一般是亚稳定的,但是,由于嵌段共聚物体系具有极其崎岖不平的自由能曲面,稳定的纳米有序结构和亚稳定的纳米有序结构之间的势垒与体系中高分子链的数密度成正比,因此,所俘获的亚稳定纳米有序结构在实验上一般都具有分钟或者小时量级的存在时间,可以采用快速降温至玻璃化转变温度以下或者使高分子链之间快速交联等方法来进行稳定。
我们分别采用组分的快速化学转变、阶跃剪切形变、快速压力变化给出了构造嵌段共聚物体系中不稳定状态的三种途径,系统研究了从这些不稳定状态出发的非平衡态结构演化动力学过程,建立了与平衡态相图完全不同的、由非平衡态结构演化动力学过程俘获的结构状态的分布图。其中,采用组分的快速化学转变和快速压力变化俘获的纳米有序结构都是亚稳定的,而采用阶跃剪切形变俘获的则都是假亚稳定的纳米有序结构(椭球和椭圆柱等),即:拥有亚稳定状态的生存时间、但存在微弱残余应力的结构。
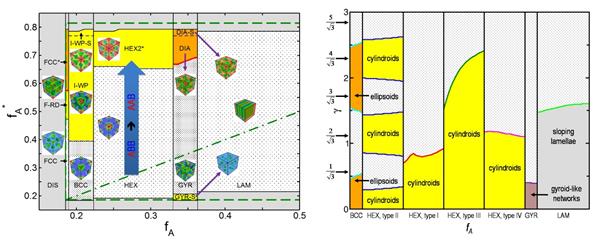
图:由非平衡态结构演化动力学过程俘获的结构状态的分布图,分别采用组分的快速化学转变(左图)和阶跃剪切形变(右图)获得,其中,左图中的fA和fA*分别表示组分的快速化学转变前后的A组分的体积分数,右图中的γ表示阶跃剪切应变的大小。
2. 聚合物非平衡态场论研究及其数值运算
基于柔性高斯链模型的聚合物自洽场理论是描述高分子复杂体系最为精确的平均场理论之一,在研究和预测高分子复杂体系的平衡态性质方面取得了十分巨大的成功。然而,由于在获取自洽场方程组时对组分空间密度分布和与组分空间密度分布相对应的势场都进行了平均场近似(正解聚合物自洽场理论),目前的聚合物自洽场理论仅仅能够获取热力学平衡状态和亚稳定状态的性质,即自由能曲面上的全局最小值点和局域极小值点,不能够获取不稳定状态的非平衡态性质。
为了能够获取高分子复杂体系中不稳定状态的非平衡态性质,我们建立了两种反解聚合物自洽场理论的高效率新数值求解技术。不同于正解聚合物自洽场理论时被研究体系的组分空间密度分布和与组分空间密度分布相对应的势场可以通过自洽迭代的方式同时进行获取,反解聚合物自洽场理论首先固定被研究体系的非平衡态组分空间密度分布,然后再采用新建立的两种迭代方法获取与组分空间密度分布相对应的势场。因此,在被研究体系的组分空间密度分布已知的情况下,采用这两种反解聚合物自洽场理论的高效率新数值求解技术可以获取相应自由能曲面上任意状态的性质,尤其是多种不稳定状态的非平衡态性质(体系的自由能和组分的化学势等),突破了传统聚合物自洽场理论仅仅能够研究热力学平衡状态和亚稳定状态的局限和计算技术瓶颈。