热增黏聚合物(TVPs)是一类功能性水溶性聚合物,属于热凝胶聚合物的范畴,近日已被国际纯粹与应用化学联合会列为2025年度化学领域十大新兴技术之一。与传统水溶性聚合物溶液在加热时黏度下降的“热变稀”行为不同,TVPs因其侧链或嵌段具备低临界溶解温度特性,可通过自缔合作用表现出独特的热致增黏现象。传统聚合物如部分水解聚丙烯酰胺在半稀溶液中主要依赖链缠结实现增黏,而TVPs在温度超过临界缔合温度后,可借助主链缠结与热响应基团间的分子间缔合协同提升黏度。然而,TVPs在低浓度下的热增黏能力及其定量机制仍需进一步探究。
近日,四川大学冯玉军团队通过“grafting onto”策略,将端氨基聚N-异丙基丙烯酰胺(NH2-PNIPAM)接枝到高分子量HPAM主链,合成了一系列高分子量的HPAM-g-PNIPAM共聚物,并系统调控了主链分子量(Mw)、接枝率(Gr)以及聚合物浓度(Cp)。通过Gompertz和Cross两种模型,建立了描述上述三参数对增黏能力影响的量化表达式。此外,基于时温叠加原理,定量比较了接枝率和聚合物浓度对接枝率与浓度对热缔合增黏的贡献程度。基于上述研究,构建了一个一个用于阐释热缔合与黏度增强之间标度关系的定量理论框架。
2025年12月2日,该论文以题为“Quantifying the Thickening Power of High-Molecular-Weight Thermoviscosifying Polymers in Pure Water”发表在Macromolecules上,四川大学博士研究生李世伟为论文第一作者,赵学之特聘副研究员和冯玉军教授为通讯作者。
本研究通过定制合成HPAM-g-PNIPAM(简称HnmGx)接枝共聚物,构建了三个系列量化体系(图1),以系统探究结构参数对热增黏行为的影响:系列一(Group 1)主链分子量不同而接枝率一致,Mw介于(5.2–16.5)×106 g×mol-1;系列二(Group 2)主链分子量相同而接枝率不同,Gr范围为1.6%–51.9%;系列三(Group 3)固定聚合物种类,改变溶液浓度Cp,区间为0.05%–0.50%。

图1.(A)氨基封端聚合物(NH2-PNIPAM)和(B)热增黏聚合物(HPAM-g-PNIPAM)的合成路径;(C)通过系统调控主链分子量(Mw)、接枝率(Gr)和聚合物浓度(Cp)构建三个样品系列,用于建立定量的结构—性能关系。
系列一:主链分子量(Mw)对热增黏行为的定量影响
随主链分子量Mw增大,HnmG50溶液在低、高温区的热剪切变稀现象逐渐减弱,中温增黏区间则明显变宽。热增黏峰值温度(Tmax)由372 K 升高至405 K,峰值黏度(ηmax)由660 mPa×s显著提升至4670 mPa×s,表明较长的主链有助于形成更稳固的PNIPAM桥联网络。基于时温叠加原理构建的时间–Mw叠加曲线显示,不同Mw体系的lnηmax高度重合,且增黏斜率一致(约为14),印证其热增黏机理相同。利用Gompertz方程拟合增黏区(Regime II)的黏温曲线发现,增黏幅度ηmax-ηass与Mw呈幂律关系(~Mw1.75),相应表达式为
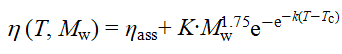
结果表明,提高主链分子量可通过促进链构象转变与网络重组,显著增强热增黏能力。
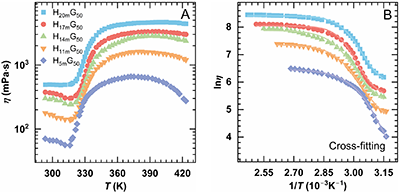

图2. 0.3% HnmG50溶液(Group 1)在不同主链分子量(Mw)下的热增黏行为:(A) 黏度随温度变化曲线;(B) Regime II 区间 lnη–1/T的Cross模型拟合;(C) 以H20mG50为参照,采用水平和竖直移位因子(aM、bM)得到的时间–Mw叠加主曲线;(D) Regime II中η–T关系的Gompertz模型拟合。
系列二:接枝率(Gr)对热增黏行为的定量影响
在主链分子量相同的H20mGx体系中,随Gr提高,体系由几乎无增黏逐渐转变为显著热增黏,表现为Tmax上升、ηmax和ηmax-ηass增大,而Tass降低,这反映了高接枝密度下PNIPAM疏水缔合占主导地位。时间–Gr叠加主曲线表明,不同Gr体系的lnηmax基本重合,但lnηass随Gr增加明显分化,且增黏斜率由2增至14。经Gompertz拟合分析,ηmax-ηass近似与Gr符合幂律关系(~Gr1.04),表达式为
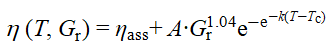
可见,提高接枝率能通过增强PNIPAM缔合与网络致密化,显著提升热增黏性能。
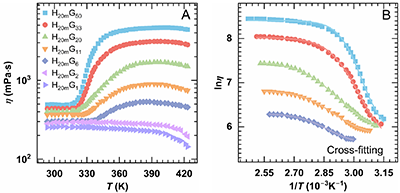
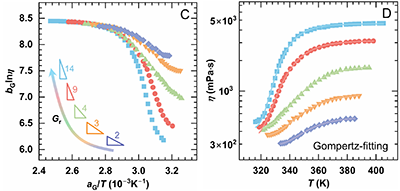
图3. 0.15%H20mGx溶液(Group 2)在不同接枝率(Gr)下的热增黏行为:(A) 黏—温曲线;(B) Regime II 区间内lnη–1/T的Cross模型拟合;(C) 以H20mG50为参照,采用移位因子aG和bG得到的时间–Gr叠加曲线;(D) Regime II中η–T关系的Gompertz模型拟合。
系列三:聚合物浓度(Cp)对热增黏行为的定量影响
以H20mG11为模型体系,当Cp从0.05%增至0.5%时,体系室温初始黏度升高,且在低至0.06 wt.%时即呈现明显热增黏。在Regime II中,随Cp增大,Tass降低而ηmax与ηmax-ηass持续增大。时间–Cp叠加曲线显示,各浓度下的lnηmax可叠合为一条曲线,而lnηass随Cp显著分化,增黏斜率由1.4提升至5.3。Gompertz拟合和标度分析后表明,ηmax-ηass与Cp近似呈幂律关系(~Cp2.01),表达式为
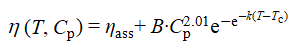
这说明提高聚合物浓度能构建更为致密的缠结与缔合网络,从而显著放大热增黏效率。

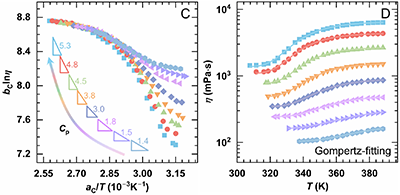
图4. H20mG11在不同聚合物浓度(0.05–0.5%)下的热增黏能力。(A)黏—温曲线;(B) Regime II 区间lnη–1/T的Cross 模型拟合;(C)以0.5%为参照,通过移位因子aC和bC得到的时间–CP叠加曲线;(D) Regime II 中η–T关系的Gompertz模型拟合。
热缔合作用对增黏能力的贡献分析
通过黏度差η -ηass表征PNIPAM热缔合对增黏的贡献,并以CPNIPAM建立标度关系。在333.15 K 参考温度下对Group 2和Group 3进行时间–温度叠加,得到bT(ηmax-ηass)曲线:在低CPNIPAM区间,两条曲线几乎重合,说明热缔合增黏贡献相近;而在高CPNIPAM下,Group 3的bT(ηmax-ηass)明显高于Group 2,对应标度指数分别为2.78 和1.10。这表明在相同PNIPAM片段浓度下,提高主链浓度(Cp)比单纯增加接枝率(Gr)能更有效地放大热缔合增黏效应。
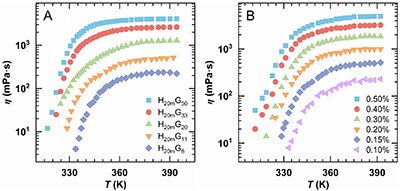
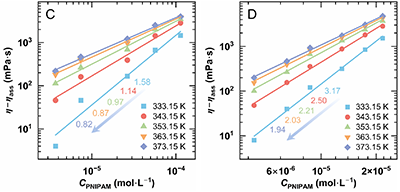

图5.(A) Group 2以及(B)Group 3的黏度差随温度的变化。(C) Group 2和(D)Group 3在333.15–373.15 K下的热缔合贡献。(E)将(C, D)中的数据经竖直移位因子bT(参考温度333.15 K)处理得到的bT(η-ηass)曲线;示意图展示微观结构演化(蓝色:HPAM主链;红色:PNIPAM)。
总结
本工作通过系统调控主链分子量Mw、接枝率Gr与聚合物浓度Cp,定量建立了TVPs的增黏能力与上述参数之间的幂律标度关系,并推导出统一的η(T; Mw, Gr, Cp)表达式。基于时温叠加分析进一步揭示,提高主链浓度相比单纯增加接枝率,能更显著放大热缔合增黏效应。该理论框架阐明了“长主链–高接枝–高浓度”协同增强热增黏效应的机制,为按需设计高效热增黏聚合物及智能流变调控体系提供了定量依据。
原文链接:https://doi.org/10.1021/acs.macromol.5c02528
下载:论文原文。
